
James Hurley
Molecular and Cell BiologyJames H. Hurley is a Professor and Judy C. Webb Chair in the Department of Molecular and Cell Biology studying protein-membrane interactions important in neurodegenerative diseases and AIDS. He received his B.A. and M.S. in Physics from San Francisco State University and his Ph. D. in Biophysics from UC San Francisco.
Spark Award Project
Activating Autophagy to Fight Neurodegeneration: Using Molecular Structure to Fight Neurodegeneration
Autophagy (literally “self-eating”) is the cell’s major mechanism for clearing out intracellular debris, and is central to maintaining health at the cellular level. Neurons are the cells that are the most vulnerable to damage when autophagy slows down in aging and disease, and a reduction in autophagic capacity is linked to neurodegenerative diseases such as Parkinson’s Disease. The machinery that carries out autophagy in cells consists of a series of protein complexes, including complexes known as “ULK1” and “PI3KC3-C1”. We are using insights into the structures of these complexes, and their dynamic changes during activation and inactivation, to develop therapies that will promote healthy autophagic function and combat neurodegenerative disease.
James Hurley’s Story
Aiding Cells’ Strategy to Survive
Missed lunch? No problem, you can just consume small bits of yourself.
As any human biology text will tell you, enzymes in the stomach and intestine break down proteins that are locked into almost every bite we eat. The proteins’ amino acid building blocks are then transported to the body’s hungry cells.

There, construction begins anew as cell machinery reassembles new proteins for whatever tasks the genes call: ramping up energy production, ferrying material to different cell sites — even switching gene activity on or off.
But cells don’t consume every protein they are offered, and leftovers can build up, clogging metabolism and threatening cell survival. Protein production can also go awry. Some must be disassembled in the cell and rebuilt, often leaving bits and pieces on the factory floor.
The Bakar Fellows Program supports research by biochemist James Hurley, professor of molecular and cell biology, to develop a new drug to boost the natural process that sweeps these threats away.
A cell’s failure to clean house poses other direct threats to survival. Over the course of its life, a cell’s machinery runs down. Mitochondria, the cell’s powerhouses, falter and free charged atoms and molecules known as oxygen free radicals to indiscriminately destroy proteins. The cell is all but doomed.
“You don’t want your cells filling up with failed mitochondria or unused protein fragments,” Hurley says.
The natural, life-saving process called autophagy cleans the table, carrying out two crucial roles at the same time. It spares the cell from multiple insults, and makes leftovers available for re-use — a boon when food is scarce.
The key player in autophagy is called — not surprisingly — an autophagosome. The autophagosome is a bubble-shaped sac that engulfs left-over amino acids, spent mitochondria and other material, and ferries them to recycling sites. An autophagosome “can fit snugly around a single mitochondrion,” Hurley says.
But as in every cell function, this too can fail. Neurons are particularly at risk, possibly due to the distance autophagosomes must travel through the cells’ long dendrites and axons to bring their cargoes back to the cell body.
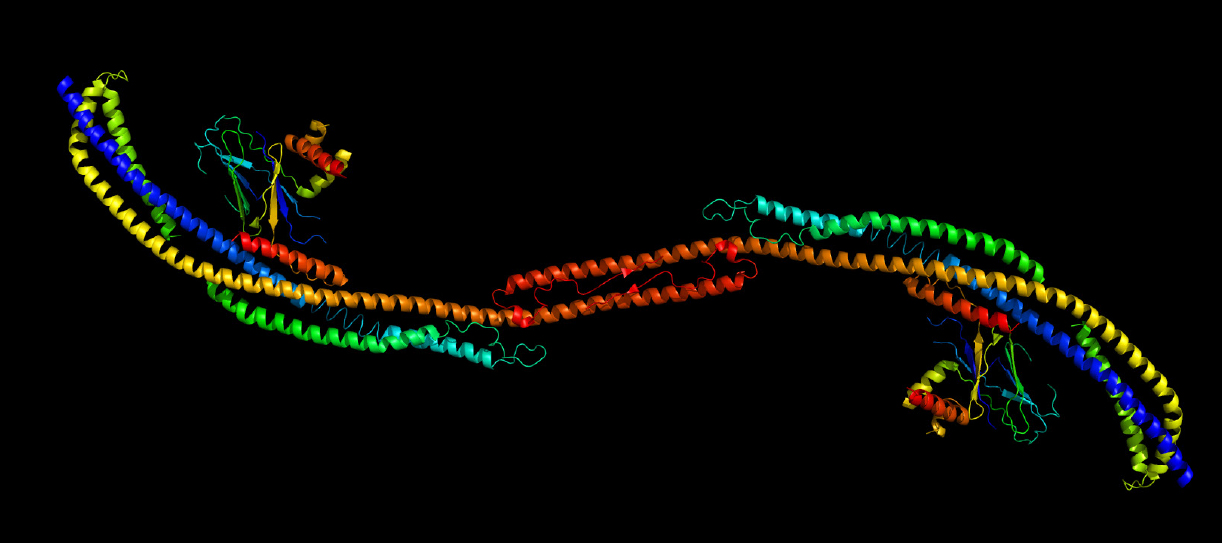
Studies in mice show that failed or sluggish autophagy causes neuron death. Inefficient autophagy may also drive the build-up of protein aggregates in neurons that is thought to cause Parkinson’s disease.
Synthesis of autophagosomes in the cell is the result of an interaction between two protein complexes — each itself made up of several proteins. Hurley’s lab has used a variety of techniques — electron microscopy, x-ray crystallography, spectroscopy and live-cell imaging — to clarify the atomic-level structure of these two units and their interaction. His research suggests that autophagosome synthesis is directly related to the distance between key sites in these two units.
The structural insights have led his lab to new research, funded by the Bakar Fellows Program, to develop a drug that can change the units’ 3-D shapes and bring them into the “activated” shape or conformation. This conformation, he thinks, would increase the cell’s production of autophagosomes.
The approach is unusual. Most pharmaceutical interest in these complexes has focused on strategies to thwart cancer growth by preventing the two complexes from becoming active — switching off autophagosome production.
“It’s much easier to turn off the signal than turn it on,” Hurley says, and the effort to do so by changing the conformation of the two protein complexes is a young field made possible by powerful structural imaging techniques.
The entire process of assembling the autophagosome takes only about ten minutes, which makes sense from an evolutionary perspective, Hurley says. Starvation can snap the cell’s autophagy machinery into action, quickly yielding nutrients to sustain cells, and allow the person — or mouse or whale — they reside in to hunt for more substantial food.
While nutrient need and the threat of spent materials drive autophagy, recent research has shown that other factors can trigger the process. Calorie-restriction diets and exercise trigger production of autophagosomes, Hurley says.
These recent, “optional” activities mimic starvation that threatened ancestors at some point, and, sadly, continue to do so in many cultures today. In societies with readily available food, autophagy’s ability to quickly provide more nutrients is far less important than its ability to clear cells of debris.
“If neurons can’t rid themselves of failing mitochondria, this defect will lead to disease, or worse,” he says. “We think we can develop a drug to reverse this threat.”